Leptin
About 15 years ago the 167 amino acid peptide hormone leptin was discovered by Jeffrey M. Friedman and colleagues through work with genetic mouse models. It is primarily expressed in adipose tissue and there is thus a close correlation between the blood level of leptin and the size of the fat tissue. A small fat tissue size, as in anorexia nervosa, correlates with low leptin levels. Since its discovery leptin has commonly been known as an appetite hormone. Because of its correlation with fat tissue size it was considered an important step in obesity research from the very beginning.
The ob/ob mice strain (the very strain that helped us isolate leptin) produce no leptin. They are extremely fat and have a voracious appetite. If injected with leptin, they eat less and loose weight. The initial results from mouse trials created an air of optimism in the obesity research area. Leptin was thought to stimulate satiety. Imagine a substance that when injected would simply turn you off food. But, as always, the human body proved more complex than first assumed. It turned out that unlike the ob/ob mouse, overweight humans often had high leptin levels. Consequently leptin injections in overweight humans, not surprisingly proved a poor treatment.

Leptin circulates in a free form and binds to leptin-binding proteins. It’s secreted in a pulsatile fashion and secretion varies with night and day. The main regulating factors for serum leptin concentration seem to be short-term food intake and the amount of energy stored in adipocytes. Although leptin correlates positively with body fat size and supposedly down regulates hunger, the overweight and obese humans still experience strong hunger. This apparent paradox sparked the term “leptin resistance”.
Leptin resistance was thought to be much like insulin resistance and thus caused by long term high levels of serum leptin. Despite high serum leptin levels, the cerebrospinal fluid/serum leptin ratio is lower in obese compared to lean individuals, suggesting that a lower central nervous system leptin transport may explain part of the supposed leptin resistance in obesity (Eckert 1998).
Local cellular hunger
Before I delve deeper into the world of leptin I need to do a quick recap. I’ve written more substantially about this before here and here, but will try to summarize the most important points (Alternatively read this, this and this).
Our sense of hunger is strongly dependant upon the state of our metabolism. For example, if we manipulate the fat tissue (by drugs or low carbohydrate diets or even exercise) into releasing larger amounts of fatty acids (energy), we don’t feel very hungry. Even more so if in addition glucose production is up regulated and glycogen breakdown is on. We also know that in most animal models, an increase in fat storage occurs prior to increases in food intake. It is thus more likely that we feel hungry and eat because we are storing fat, than it is that we are storing fat because we feel hungry and subsequently eat more.
Of course we don’t feel very hungry when we have a high fat oxidation. As humans we get the energy we need to sustain life and locomotion from two sources; food or stores in our body. When the stores provide a larger part of the energy needed the need for acquiring it from food declines. Hunger declines, metabolism is turned up and energy stores in our body shrink.
If, we on the other hand manipulate the fat tissue in the direction of storage, for example through increasing insulin levels we shut down fat release from adipose tissue. This effect takes place whether the insulin comes from injections or your own pancreas. The energy provided by the body stores is no longer enough to sustain high metabolism. The result is increased hunger, lower body temperature and no drive to exercise (fatigue). Many overweight people recognize these symptoms and meet them at a daily basis. These are the symptoms of a hungry body in a storage mode. It is hungry because much of the energy it needs is stored away. In this mode, if we don’t eat and don’t fill the energy gap with energy from food, the body will starve. It will starve no matter how much energy we’ve got stored as fat. What matters is if or how much of the energy stores are available for use.
Starving the body (even if you are overweight) will cause a great deal of things. In females, amenorrhea or loss of menses, the shutting down fertility is common. Fertility (and sex drive) is one of the first things to go when the body feel its starving. The simple explanation for why an energy deficit causes disruption of the reproductive function is that reproductive function has a low priority in the survival of mammals. Functions essential for survival are those of basic cellular maintenance, keeping correct body temperature and locomotion to obtain food. These functions are maintained at the expense of other functions (e.g. reproduction).
In the words of George Wade et al (1996):
” …it is worth noting that the low priorities of both reproduction and fat storage vis-a-vis processes necessary for survival could account for their habitual association. Exercise, exposure to low temperatures, excessive fat storage, or poorly controlled diabetes mellitus illustrate this second point.”
Take a strong note of the “excessive fat storage” part. Fat storing is from the body’s view not necessarily considered a state of energy surplus, but often that of energy deprivation. What is deprived, are all the tissues of the body, and even fat tissue itself. For as long as energy is being locked into the form of triacylglycerols and the hormonal environment hinder its release, no tissues can use it.
In animal studies, feeding a high-fat diet (which increases the energy flow from fat tissue to other tissues) may ameliorate reproductive deficits. Energy deficits resulting from inadequate energy intake are also more extreme when consuming a high carbohydrate diet.
Decreased reproductive function is but one symptom of starvation. When deprived of energy, even muscles may brake down to a larger extent to supply glucose for fuel. Supporting this theory are observations of sudden increases in muscle mass in ketogenic diet studies, and findings that the muscles in obese woman act very much like muscles in a starving person (Hittel 2009).
The major point here is that energy availability of the whole body does not reflect the energy availability of specific tissue cells. And hunger is largely the result of our metabolism, the regulatory point seem to be the production of ATP in liver cells (Friedman 1999).
Uniting the theories
Now for how leptin relates to hunger and our metabolism.
Leptin as an energy flux indicator
Leptin was thought to be a satiety signal. But, according to resent research, leptin’s main function is not simply to directly regulate hunger and satiety, but to inform the organism that there is enough energy to sustain life. The major physiologic role of leptin seems to be to signal available energy to the hypothalamus.
Increasing leptin increases fat oxidation. The finding is of great importance because increasing fat oxidation by any means, reduce hunger. Arch et al probably hit the nail on its head when they proposed that leptin is not raised in obese individuals because of leptin resistance, but because leptin is opposing other forces that promote obesity. Because of the opposing forces that drive fat storage, leptin is desperately trying to get the energy stored as fat transported to other tissues.
In anorexia nervosa leptin levels are low, very low (Eckert 1998). The oxidation of stored fuels is kept at a minimum and consequently the body is no longer signaling that it has energy surplus, which it hasn’t. It needs energy from food.
When fasting, leptin levels decrease rapidly before and out of proportion to any changes in fat mass, thus likely signaling an energy gap. A gap existing because the oxidation of stored fuels is not in itself enough to keep metabolism high. Consequently the body need energy from food and lack of leptin is signaling just that.
Expression of leptin from adipocytes is directly related to the glucose uptake by adipocytes. Glucose uptake is directly related to insulin level, and glucose level is directly related to fat oxidation. When fat oxidation is low, as with a high carbohydrate diet, the body relies heavily on glucose for fuel. The strong reliance on glucose increase the probability of low glucose levels with consequent decreased leptin secretion and increased hunger.
Ketones are produced to spare glucose. Most of the cells that can metabolize glucose can also metabolize ketone bodies. When people are fasting they commonly experience a great hunger the first days, but as glucose level drops, fat metabolism is turned up and keton production is increased. When ketone body production is increased, hunger declines, even though a person is still fasting. Thus hunger is controlled by the total rate of oxidation of fuels and not by the amount of energy ingested.
The same mechanism comes into play on a low carbohydrate diet when insulin is decreased and ketone body production is increased (Johnstone 2008, Boden 2005).
When fat oxidation increases, whether it is by eating less (calorie restriction) or specifically reducing glucose and insulin load (low carbohydrate diet), leptin decreases. But this may not mean increased hunger. There may no longer be a need to overpower other fat storing effects. A decreased sensation of hunger may actually appear simultaneously with decreased leptin levels, further supporting the notion that leptin is not simply a hunger signal.
Leptin is decreased with dieting also because total fat mass is decreased. But, leptin concentration decrease after weight loss has been found to be disproportionate to changes in adiposity. These observations suggest that other factors in addition to adipose mass modulate leptin secretion. On low calorie diets, the individuals who experience the greatest increase in hunger, and therefore those who probably have the lowest fat oxidation rates, are also those who have the larges decrease in leptin (Keim 1998).
When the overweight person is hungry, this seems a paradox to those only preoccupied with total body energy expenditure and intake. It is not a paradox. It is a completely natural response to specific tissues starving because too much of the energy ingested is stored in the adipose tissue. The overweight person may also have a high level of leptin in combination with high level of hunger, because some tissues are in fact starving. Leptin is perhaps increased in this condition to increase energy availability, but does not in itself down regulate hunger. Hunger is reduced only if fat oxidation is properly increased.
In one study (Cooling 1998) the researchers found that subjects habitually consuming a high-fat diet had raised leptin concentrations and a higher basal metabolic rate (BMR) than subjects with the same BMI and adiposity habitually consuming a low-fat diet. In this case leptin seem to be high in the individuals on the high fat diet because it signals an energy surplus. A high fat intake leads to less fat storage than do high carbohydrate intake. The finding also shows how diet and thus metabolism influences leptin secretion independent of fat tissue size.
Leptin resistance
Evidence for leptin resistance was first based solely on the finding that obese humans generally have elevated serum or plasma leptin concentrations compared to lean subjects (arch 1998). Because the theory was not supported by experimental data, Arch et al (1998) proposed that that leptin concentrations are not raised in obese individuals because of leptin resistance, but because leptin is opposing other forces that promote obesity.
But does leptin resistance really exist? Or can the findings which lead to the theory perhaps be explained through the local cellular hunger – energy oxidation hypothesis? The question proves difficult to answer. In 1998, roughly two years after the discovery that overweight individuals had high levels of leptin, some researchers already considered this hypothetical resistance to be nonexistent.
There still are several indications that a form of leptin resistance might exist. For one there is the fact that cerebrospinal fluid/serum leptin ratio is lower in obese compared to lean individuals. Although, this may just be a transport problem not related to an actual resistance. Several rat studies have shown increased fat oxidation rates by skeletal muscles when exposed to leptin. In humans however it is not quite that easy. One study (Steinberg 2002) showed a greatly increased fatty acid oxidation in muscles from lean individuals in vitro. Muscles tissue from overweight individuals however, did not show an increase when exposed to leptin. The finding is of course considered to be an indication of leptin resistance in the overweight, but remember these cautioning word from the authors; “it should also be noted that the non physiological conditions imposed in such a preparation (i.e., high leptin, absence of insulin and other hormonal factors) make it difficult to extrapolate our findings to the in vivo condition.”
Also, animal studies have demonstrated that 4 weeks of high fat feeding can induce leptin resistance in skeletal muscle, as demonstrated by the elimination of leptin’s stimulatory effect on fat metabolism. If the stimulatory effect is gone we do indeed need to call it a resistance.
Leptin may be increased in obese individuals and so representing an attempt to overpower fat storing processes as well as possibly representing a leptin resistance. When weight is lost there seem to be a decrease in leptin either because of a reduced need for its effect on fat metabolism or because of an increase in leptin sensitivity. In any case, a high level of leptin in overweight do not likely cause overweight. It is there to reduce fat storage rather than to drive it.
Blüher et al (2009) argues that leptin resistance or hyperleptinemia causes not only an increasing degree of obesity, but is also associated with increased lipid storage in muscle, liver, and other tissues, dysfunction of several neuroendocrine axes, including the reproductive, thyroid, and adrenal axes, as well as abnormal function of the immune and autonomic system i.e. thermoregulation, energy expenditure, and others. Looking at this cluster of symptoms, we are looking at any metabolic resistant overweight person. All symptoms can be easily explained without leptin and it is thus unlikely that leptin play the causal role.
Uniting
Leptin was originally believed to control hunger, although it was never actually clear if it “controlled” it, or was simply a part of the intricate physiological interaction that is hunger. Leptin was later discovered to also be a part of the regulation of reproduction and the hypothalamic-pituitary-gonadal (HPG) axis. Animals that lack leptin (ob/ob mice) or have obvious leptin resistance (db/db mice and fa/fa rats) fail to achieve puberty and are infertile.
This is where we most easily see the theories unite. Reproduction is, as Wade shows us, highly sensitive to fluctuations in energy as is leptin. Leptin acts as a signal informing the different tissues of the energy state of the body, rather than itself controlling the energy metabolism.
The features of hypothalamic hypogonadism in women (low levels of sex hormones) and it’s associated disturbances can be restored by leptin administration. The question is, do we simply trick the body into believing there is more energy than there actually is, or do we actually increase energy availability?
Amenorrhea, the lack of menses, may also be induced by exercise. Endurance trained athletes have a higher prevalence of amenorrhea or dysmenorrhea than non exercising controls. It is normalized with leptin injections. Administering leptin may increase fat oxidation and thus, as Friedman et al has reported, also increase hepatic ATP levels and by doing so may restore fertility function.
Both increased levels of leptin, as in obesity, but also low levels act inhibitory in the HPG axis. Blüher el al claims that: “These results underline a pivotal role of leptin in regulating reproductive function and strengthen the hypothesis that leptin is one of the factors mediating reproductive abnormalities in several disease states.” But it may not be this way. Leptin is not necessarily “controlling” fertility. It may simply be the messenger informing the body of its energy status. Reproduction is regulated in accordance with energy status.
Blüher et al continues: “We have shown that leptin may serve as a signal to convey information to the reproductive system that the amount of energy stored in the body as fat is adequate not only for the survival of the person but also for carrying a pregnancy to term.”
This claim however, rests on the assumption that the energy stored as fat is available for use. This is not always the case.
When leptin resistance seem to be causing obesity in rodent models and the rare cases in humans, it is likely because the different body tissues and the hypothalamus constantly experience an energy shortage. Metabolism with all its manifestations, fertility included, is adapted. The body does what it can when it senses energy to be in short supply. It increases hunger, decreases the metabolic rate and effectively shuts down unnecessary processes.
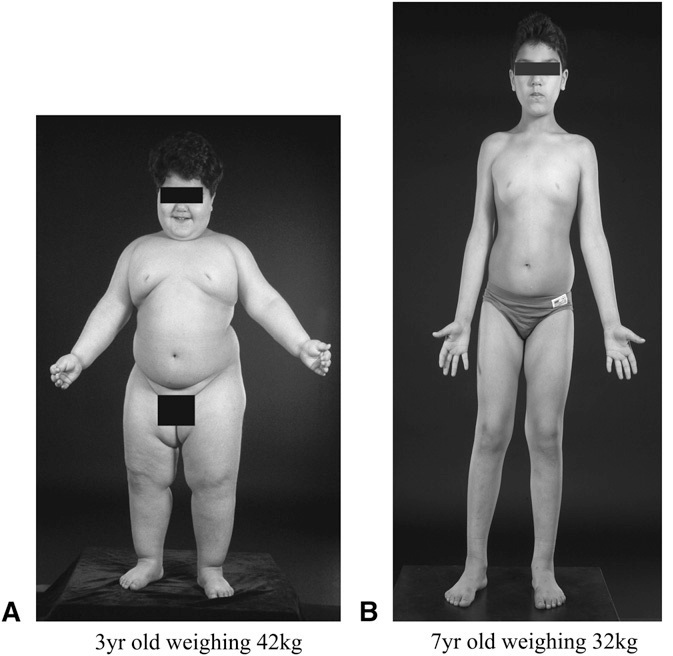
Effects of recombinant human leptin treatment in a patient with congenital leptin deficiency. (A) Before treatment. (B) After treatment. From: Leptin: a pivotal regulator of human energy homeostasis, Farooqi 2009.
States of congenital leptin deficiency because of mutations of the leptin gene have been associated with severe obesity, glucose intolerance, and insulin resistance in humans. All expected symptoms if fat oxidation is low.
Despite the initial hopes, placebo-controlled trials in obese persons over periods of several weeks with leptin-treated subjects have not shown impressive results on weight loss. This is not surprising, considering that many obese persons have high leptin levels, high glucose level and high insulin resistance, and thus a fat tissue which reluctantly gives away energy. A slight increase in fat oxidation with leptin replacement therapy is expected to reveal nothing but small decreases in fat tissue, as long as nothing else is done with the fact that the tissue is in a storage mode.
Even in patients with severe lipodystrophy (inability to store fat/loss of fat tissue) leptin replacement decreases fat mass. The decrease in fat mass is an indicator that fat oxidation is increased and in accordance symptoms of starvation are improved (Oral 2002). At least in the short run. Women with anorexia nervosa and with cachexia resulting from cancer or severe chronic infections also show many of the symptoms of a starving body, including low leptin levels. Despite the fact that leptin would have the highly undesirable effect of inducing weight loss in these patients leptin treatment is still recommended (Friedman 2009). It is another example of a treatment aimed at ameliorating symptoms, without positively (probably negatively) effecting the underlying cause.
Control by food intake
Seen together the current data point to leptin as a signal informing the hypothalamus of the availability of oxidizable fuels.
If leptin is secreted in response to the energy metabolism, we would expect macronutrient intake to influence leptin level. Several studies have found such an effect, while others have not.
An increase in dietary protein from 15% to 30% of energy at a constant carbohydrate intake has produced sustained decreases in caloric intake, hunger and leptin levels (Weigle2005).

Havel et al (1999) reported acutely decreased leptin concentration after ingestion of a high fat, low-carbohydrate diet. Jensen et al (2006) found that persons in the highest quintile for whole-grain intake in a prospective study had 11% lower circulating leptin compared to those in the lowest quintile. It is intriguing to think that lower in this case could mean increased hunger response due to lower fat oxidation, but we must consider the possibility that it simply reflects and reduced leptin resistance. Volek et al reported a 42% decrease in leptin with a low carbohydrate diet (12%CHO) and 18% decrease in leptin with low-fat diet (24% fat) for 12 weeks in overweight subjects. Once again, the results could indicate a lack of energy availability or improvement in leptin resistance.
But, because we know that carbohydrate restriction greatly increases fat oxidation, reduces hunger and may even increase heat production and locomotion, it seem most likely that the larger decrease in leptin on a low carbohydrate compared to a high carbohydrate diet represents a decreased need to overpower fat storing mechanisms. The significant decrease in leptin found by Volek et al persisted after normalization of body and fat mass.
Expression of leptin from adipocytes is directly related to the glucose uptake by adipocytes. This could in itself explain why leptin is reduced more with carbohydrate restriction. Leptin also decreases with decreasing weight, because fat tissue is the main secreting organ. This means that in an overweight person with hyperleptinemia a reduction in leptin level is expected with weight loss and is not negative just because high leptin may reduce hunger.
It seems that increases in leptin usually signal a surplus of energy, but not in the obese. In the obese the high levels seem to be caused by the body’s desperate attempt to increase fat oxidation.
In support of this are the findings that injections of leptin increase fat oxidation in combination with reduced food intake. That dieting which increase fat oxidation cause reduced levels of leptin in the overweight.
Summing it up
Believing that leptin may prove to be an important treatment for obesity, trough down regulating hunger, is nothing short of crazy. There is little doubt left in the literature that the hunger experienced by an overweight person, stems from the unavailability of oxidizable fuels. Hunger is not what must be improved, rather we must increase the release and oxidation of the fat situated in adipose tissue stores, and hunger will decrease.
Although leptin is expressed in relation to adipose tissue mass, its main function is not so signal adipose tissue size, but fluctuations in available oxidizable fuels. When leptin treatment ameliorates adverse symptoms related to conditions of low fat mass, i.e. anorexia nervosa or lipodystrophies, the mechanism is likely an increase in fat oxidation and thus a small increase in oxidizable fuels. Treating these conditions with leptin is counterintuitive and may even cause adverse effects over time. Blaming leptin is equivalent to killing the messenger.

Legg igjen en kommentar